
In the pharmaceutical industry, the development of a new drug compound is a rigorous and intricate process. One of the critical stages in this journey is the method development phase, where high-performance liquid chromatography (HPLC) plays a pivotal role. This case study explores the systematic approach taken for the method development of a novel anti-inflammatory drug compound, which we will refer to as Compound X. The goal was to establish a robust and reliable HPLC method for the analysis of Compound X in various formulations and biological matrices.
Background
Compound X was designed to target a specific inflammatory pathway, showing promising results in preliminary studies. As the compound moved into the preclinical stage, there was an urgent need to develop a reliable analytical method for quantifying the drug’s concentration in formulations and biological samples. A suitable HPLC method would ensure quality control and facilitate pharmacokinetic studies necessary for understanding the drug’s behavior in the body.
Objectives
The primary objectives of this method development project were:
- To develop a validated HPLC method for the quantitative analysis of Compound X.
- To assess the method’s robustness and reproducibility under various conditions.
- To establish a standard operating procedure (SOP) for routine quality control.
Method Development Process
1. Preliminary Research
Before commencing the method development, a comprehensive literature review was conducted. This included previous studies on similar compounds, existing HPLC methodologies, and the physicochemical properties of Compound X. Key characteristics such as solubility, stability, and potential degradation pathways were evaluated.
2. Selection of HPLC Conditions
Column Selection
Based on the preliminary research, several columns were considered. A C18 reversed-phase column was selected for its versatility and effectiveness in separating non-polar to moderately polar compounds.
Mobile Phase Optimization
Initial experiments indicated that Compound X had moderate polarity. Thus, a gradient elution method was proposed using acetonitrile and phosphate buffer (pH 7.0). The mobile phase composition was fine-tuned through a series of trials, adjusting the ratio of organic solvent to buffer to achieve optimal resolution and peak shape.
Flow Rate and Temperature
The flow rate was initially set at 1.0 mL/min, with the temperature maintained at 30°C. These parameters were adjusted based on preliminary results to ensure good separation and retention times.
3. Sample Preparation
The analysis required careful sample preparation due to the potential for matrix effects. A solid-phase extraction (SPE) technique was employed for biological samples to remove interfering substances. For formulation samples, dilution and filtration were sufficient to prepare the samples for HPLC analysis.
4. Validation of the Method
Once the initial method was developed, it underwent rigorous validation according to the ICH Q2(R1) guidelines. The following parameters were assessed:

Specificity
Specificity was evaluated by analyzing the samples in the presence of potential excipients and metabolites. The method demonstrated clear separation of Compound X from all interfering substances.
Linearity
Linearity was assessed across a concentration range of 10–1000 ng/mL. A calibration curve was generated, yielding a correlation coefficient (R²) of 0.999, indicating excellent linearity.
Accuracy and Precision
Accuracy was evaluated by comparing the measured concentration of Compound X against known standards. Precision was assessed through intra-day and inter-day variations. The relative standard deviation (RSD) was found to be within acceptable limits (< 2%).
Limit of Detection (LOD) and Limit of Quantification (LOQ)
The LOD was determined to be 2 ng/mL, and the LOQ was found to be 10 ng/mL, making the method suitable for detecting low concentrations of Compound X in biological matrices.
5. Robustness Testing
To ensure the method’s robustness, variations in key parameters—such as mobile phase composition, flow rate, and column temperature—were tested. The method showed consistent performance with minimal changes in retention time and peak area, confirming its reliability for routine analysis.
Results
The developed HPLC method for Compound X demonstrated high sensitivity, specificity, and reproducibility. The retention time for Compound X was approximately 7.55 minutes, providing a clear and well-defined peak.
Example Chromatogram
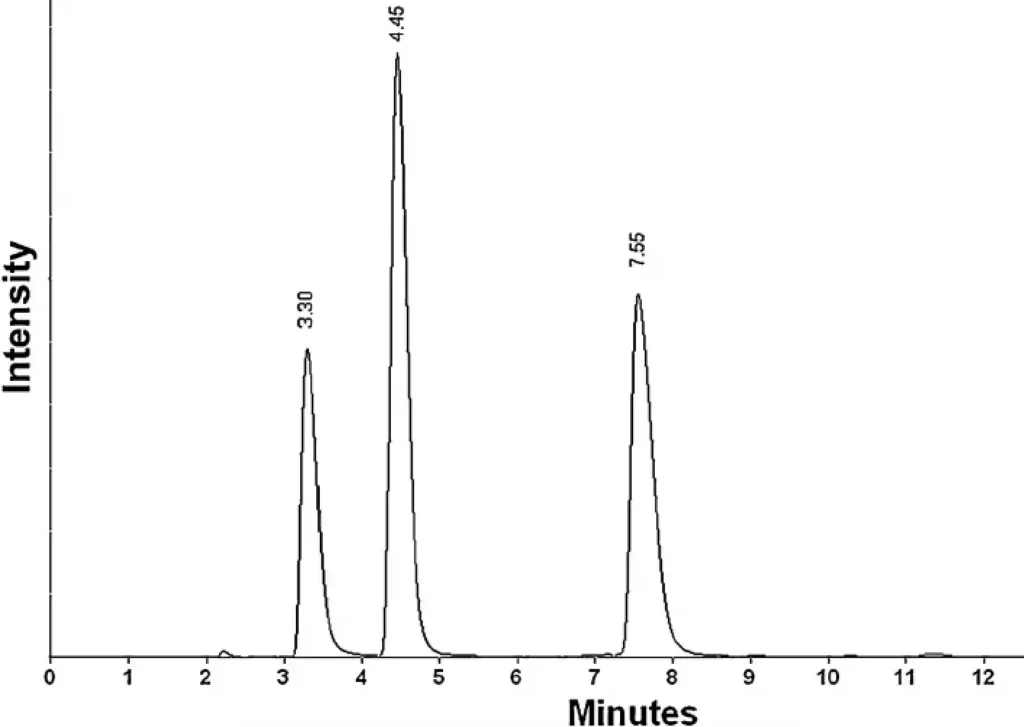
Figure 1: Chromatogram of Compound X demonstrating clear separation from impurities and excipients.
Application in Pharmacokinetic Studies
With the validated HPLC method in place, pharmacokinetic studies were conducted using the method to analyze blood plasma samples from preclinical trials. The data obtained provided valuable insights into the drug’s absorption, distribution, metabolism, and excretion (ADME) profile.
The results indicated that Compound X had a half-life of approximately 4 hours, with peak plasma concentrations occurring at around 1 hour post-administration. This information was critical for determining dosing regimens for subsequent clinical trials.
The established method not only meets regulatory requirements but also supports ongoing research into Compound X’s therapeutic potential. Moving forward, this method will be instrumental in ensuring the quality and efficacy of Compound X as it progresses through the clinical trial phases.
Future Directions
As Compound X advances, further method refinements may be necessary to accommodate different formulations and delivery routes. Additionally, exploring alternative chromatographic techniques, such as ultra-high-performance liquid chromatography (UHPLC), could enhance sensitivity and throughput. Continuous monitoring and adaptation of the analytical method will be essential to meet the evolving needs of the drug development process.


